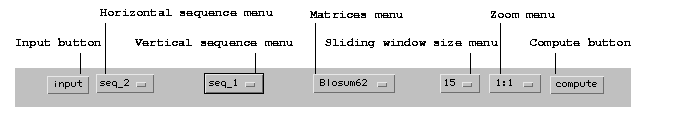
dotlet menu bar
dotlet program. For a quick summary of
dotlet's less obvious commands, see the command summary page. There is
also an examples page that
shows you the kind of features that can be discovered
with this program.dotlet's menu bar:|
|
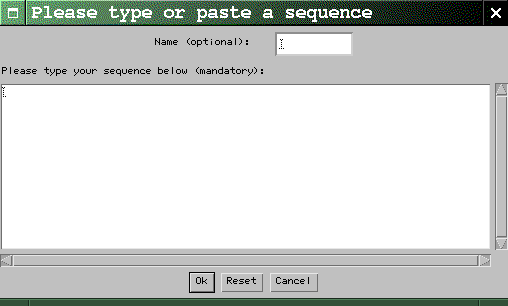
|
dotlet
will create one for you. When you're done,
click the Ok button. Characters
other than letters are ignored, so it is ok
to cut-and-paste a sequence that has spaces
or position numbers in it. Case is not
important.

|
|
|

|
|
<'
(up left), '>' (down right), '['
(up right), and ']' (down left). When the
display is zoomed, the cursor will not be updated at
every keystroke, but the alignment window will.
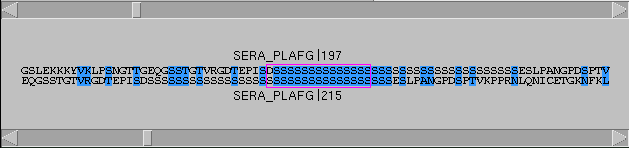
dotlet
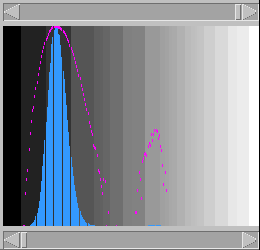
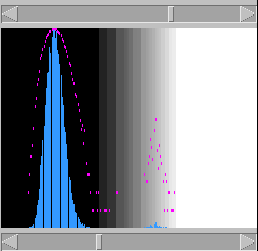
will reverse complement one of the sequences and perform
a second comparison. Each pixel is set to the best of
these two scores. This enables to see structures like
stem-loops, for example. The alignment window will also
display the reverse complemented sequence.